Story
SubQuad: Near-Quadratic-Free Structure Inference with Distribution-Balanced Objectives in Adaptive Receptor framework
Key takeaway
A new algorithm can efficiently analyze immune system data to identify rare but important immune cell types, helping doctors understand immune responses and diseases.
Quick Explainer
SubQuad introduces an end-to-end framework for scalable and equitable analysis of large immune repertoires. It overcomes the quadratic cost of pairwise affinity evaluations through an antigen-aligned MinHash indexing approach. The pipeline integrates a multimodal fusion backbone that captures both fine-grained sequence edits and higher-level biochemical structures. Crucially, it employs a fairness-aware spectral clustering objective to ensure proportional representation of rare, clinically significant clonotypes, addressing dataset imbalances that can obscure minority populations. By aligning computational objectives with biological realities, SubQuad offers a principled approach to large-scale immunoinformatics, enabling applications such as epitope prioritization, biomarker discovery, and vaccine design.
Deep Dive
Technical Deep Dive: SubQuad - Near-Quadratic-Free Structure Inference with Distribution-Balanced Objectives in Adaptive Receptor Framework
Overview
SubQuad is an end-to-end pipeline for scalable, antigen-aware, and equity-preserving analysis of large immune repertoires. It addresses two key challenges in comparative analysis of adaptive immune repertoires at population scale:
- The near-quadratic cost of pairwise affinity evaluations
- Dataset imbalances that obscure clinically important minority clonotypes
The pipeline integrates three key innovations:
- An antigen-aligned MinHash retrieval module for near-subquadratic candidate reduction
- A multimodal fusion backbone with a differentiable gating controller to capture both fine-grained edits and higher-level biochemical structure
- A fairness-aware spectral clustering objective with automated equity calibration to ensure proportional representation of rare antigen-specific clonotypes
Problem & Context
- Immune repertoires commonly comprise millions to hundreds of millions of distinct receptor sequences
- Comparing repertoires across individuals or clinical states can reveal antigen-specific response patterns that inform vaccine design, guide cancer immunotherapy, and support autoimmune disease monitoring
- However, pairwise affinity evaluations grow quadratically with the number of sequences, and naive comparison becomes infeasible for modern datasets
- Many scalable pipelines process receptor sequences as generic strings, discarding antigen-relevant signals important for epitope binding
- Subgroup representation has received limited consideration, risking systematic omission of low-prevalence but clinically consequential clonotypes
Methodology
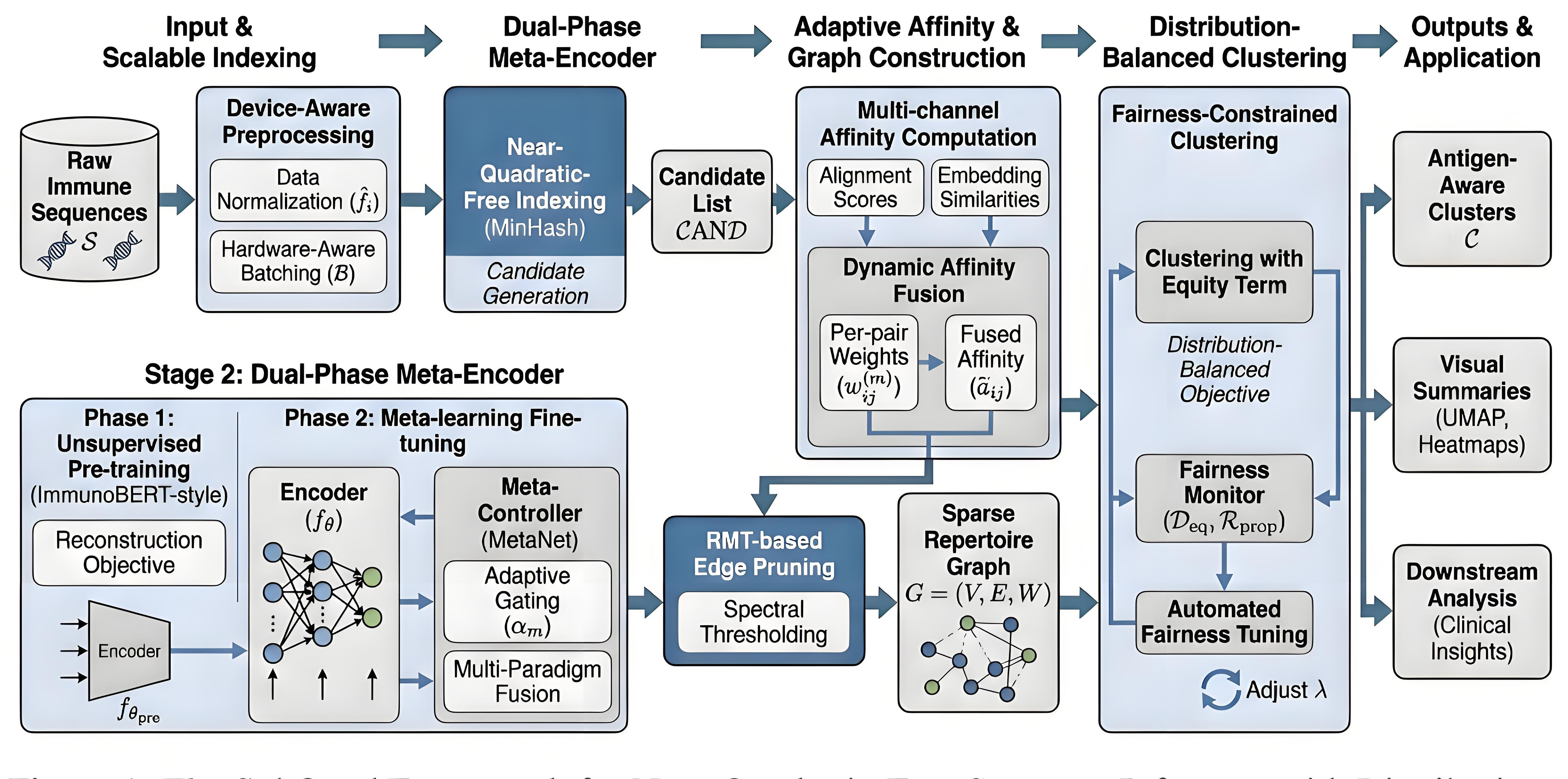
Scalable Preprocessing
- Raw sequences $\mathcal{S}$ are processed via MinHash-based Indexing to generate a sparse candidate list $\mathcal{CAND}$ and optimized using hardware-aware batching $\mathcal{B}$
Representation Learning
- A Dual-Phase Meta-Encoder utilizes ImmunoBERT-style pretraining followed by MetaNet fine-tuning
- The Meta-Controller dynamically adjusts gating weights $\alpha_{m}$ for multi-paradigm fusion
Graph Construction
- Multi-channel affinities are integrated via Dynamic Affinity Fusion to produce $\widetilde{a}_{ij}$
- This similarity matrix is refined through RMT-based Thresholding (eigenvalue spectrum analysis) to produce a sparse weighted graph $G=(V, E, W)$
Fairness-Constrained Clustering
- The graph is partitioned into clusters $\mathcal{C}$ by optimizing a joint objective of spatial cohesion and Jensen-Shannon Equity
- An Automated Fairness Tuner dynamically calibrates the trade-off weight $\lambda$ to meet target disparity $\delta_{\max}$
Data & Experimental Setup
- Datasets: VDJdb, McPAS-TCR, NEPdb
- Evaluation metrics: Throughput, Recall, Memory, Purity, Equity Score
- Hardware: Single-node GPU (dual A100s), Distributed cluster (8 T4 nodes), Heterogeneous CPU-GPU-FPGA
Results
Optimized Indexing Mechanism
- Antigen-aware MinHash LSH index with block-aligned storage achieves 58% reduction in memory consumption compared to FAISS
Query Processing Efficiency
- Sub-millisecond median latencies under high concurrency through NUMA-conscious memory partitioning, lock-free coordination, and multiversion isolation
Component Impact Analysis
- GPU parallelism yields 67% throughput gains
- Equity-aware objectives improve cluster purity by ~16% compared to fairness-excluded variants
- Embedding-only pipelines trade memory efficiency for lower throughput and reduced purity
Immunological Performance and Robustness
- Enforcing fairness via Demographic Parity reduced subgroup representation bias from 20% to 12% in the tumor neoantigen setting
- Equalized Odds improved subgroup-balanced recall in viral epitope classification
Scalability Evaluation
- Processing 1 million sequences in under 40 minutes on a single node
- At 1 million sequences, SubQuad achieves recall@100 ≥ 0.96 vs. 0.92 for the MinHash-only baseline
Interpretation
- SubQuad provides a scalable and biologically valid graph-learning platform for epitope prioritization, biomarker discovery, and vaccine design
- The fairness constraints are grounded in immunological principles, ensuring that rare but clinically significant clonotypes are not overlooked
- By aligning computational objectives with biological realities, SubQuad offers a principled approach to large-scale immunoinformatics
Limitations & Uncertainties
- Verification of runtime and memory claims depends on complete reporting of index and kernel configuration details
- Long-term evaluation of the framework's clinical impact requires further validation in translational studies
What Comes Next
- Extend SubQuad to model longitudinal repertoire dynamics
- Incorporate epitope- and phenotype-supervised representations
- Evaluate privacy-preserving federated learning across multi-center cohorts